Title: Midlife Hypertension and Alzheimer Disease: a Meta
analysis
Charles
L. Carter, PhD; Melanie Wiebe PT (2002)
updated with 6 figures
California
State University, Long Beach
Keywords: Midlife Hypertension, Alzheimer Dementia, Meta
analysis,
Vascular Factors
Corresponding Author: Charles L. Carter, PhD
Abstract:
The result of the several studies of
midlife hypertension (mHTN) and Alzheimer Disease
(AD) have not bee
laid out in a convincing manner.
Methods. Studies that were similar in design were selected. The papers
were then judged for inclusion in the
study based on: 1) longitudinal study of three or more years 2) measure
of dementia via DSM-IV criteria, the
MMSE, the 3MSE, the CASI, or the NINCDS-ADRDA criteria and 3)
published in a major journal. Results.
Five studies were found that could be included in the analysis.
The combined odds ratio of 5 dementia
studies was 1.09 suggesting hypertension is not a factor when all
are lumped together. However the odds
ratio of all AD studies of both elevated midlife diastolic (>9otorr) an
elevated systolic (>160 torr) are
much greater than 1.0 with a combined odds ratio of 1.84 suggesting that
midlife hypertension persists as a
factor in AD. Conclusions. The possibility exists that AD has both vasculature
and neurological precursors and the
elements that could tie them together are discussed.
Short title: Midlife
Hypertension and Dementia
Keywords: Midlife
Hypertension, Alzheimer Dementia, Meta analysis, Vascular
Factors
INTRODUCTION
The link between hypertension and AD
has been elusive. The apparent inability of
presently available drugs to alter the
course of AD could be a signal that it is time to
change the way we think about AD and
its therapeutics [1]. Many extensive studies
have been undertaken however the data
of the several studies have not been laid out in a
convincing manner. A major problem is
that the abnormal presentation of blood pressure
is far more complex than envisioned
[2]. The fact that we are struggling to demonstrate a
physiological effect that can take
many paths in spite of the relatively rigid changes in
anatomy
are reminiscent of causal relationship problems of CHD and essential
hypertension. The goal of Meta
analysis is to standardize the differences between
treatment groups (effect size) by
standardizing different but conceptually related studies,
allowing the comparison of dependent
variables [3]. This analysis is a presentation of
previous longitudinal studies
normalized in a way that would allow a fair comparison.
Dementia, and specifically Alzheimer's
disease (AD), has been viewed as
primarily a neural disease but most
studies have been unable to rule out vascular risk
factors [4-9]. There is a controversy
in the literature regarding the relationship between
midlife hypertension and dementia
[10,11]. The purpose ofthis meta-analysis is to
compare these studies and allow the
reader to draw conclusions regarding the
relationships between dementia and
hypertension and AD and hypertension.
While some suggest that vascular
factors are associated solely with vascular
dementia as opposed to AD [12]. Recent
literature however shows that vascular factors
may affect AD as well [13-15]. Launer et al conducted a prospective study of37354
subjects from Oahu,
Hawaii who were part of the Honolulu Asia Aging Study (HAAS)
[4]. Participants
had their blood pressures measured four times between 1968 and 1991,
at which time they
took the Cognitive Abilities Screening Instrument (CASI). The CASI
is a composite of the Hasegawa Dementia Scale, the Mini
Mental State Examination
(MMSE) and the
Modified Mini Mental State Examination (3MSE). It
has been
validated as a
screening tool for dementia with 80% sensitivity and 77% specificity. The
MMSE is a screening
tool for dementia with 87% sensitivity and 82% specificity. The
results of Launer's study showed that midlife diastolic blood pressure
is not associated
with the development
of dementia while midlife systolic blood pressure is associated with
the development of
dementia. Kivipelto et ai,
studied 1400 participants who were
randomly selected
from two other trials [7]. The mean follow-up time was 21 years and
participants were diagnosed
with AD using the National Institute of Neurological and
Communicative
Disorders and Stroke-AD and Related Disorders Association (NINCDSADRDA)
criteria [16]. The
investigators found that increased systolic blood pressure in
midlife was a
significant risk factor for AD in later life. Guo et al measured blood
pressure and
cognitive performance of 1736 participants aged 75-101 over a period of
40.5 months [17].
Cognitive performance was measured by the MMSE and cognitive
impairment was
considered a score of less than 24 on the MMSE. Guo et al found that
systolic
hypertension is positively related to cognitive performance and that low SBP
predicts poor
cognitive function [17]. Morris et al, studied 378 subjects between 1973
and 1988 in a
longitudinal cohort study in east Boston [6]. They diagnosed AD 13 years
after initial blood
pressure measurements using NINCDS-ADRDA criteria. Morris et al
found no association
between blood pressure measured 13 years before and AD.
Petrovich
et al studied 210 dead participants from the HAAS [5]. The researchers
compared midlife
blood pressure and later life dementia onset. They also studied the
number of neuritic plaques (NP) and neurofibrillary tangles (NFT) in
the patients but
made no further
diagnosis of AD other than the clinical diagnosis already given to the
patient before
death. Petrovich et al, found a positive correlation
between high midlife
systolic and
diastolic blood pressure and NP and NFT [5].
METHODS
The methods for this
meta-analysis involved five steps. First, all studies about
longitudinally
measured blood pressure and later development of dementia were gathered
through an
exhaustive search ofpublished literature using
Medline, PubMed, the
Cochrane
Collaboration, Cochrane Controlled Trials Register, National Research
Register,
ClinicaITrials.gov, and references from relevant articles. The papers were then
judged for inclusion
in the study. The inclusion criteria were: 1) longitudinal study of
three or more years
2) measure of dementia via DSM-IV criteria, the MMSE, the 3MSE,
the CASI, or the
NINCDS-ADRDA criteria and 3) published in a major journal. Five
studies were found
that could be included in this present analysis [4-7,17].
Four relevant studies
were excluded from this analysis. Elias et ai, as part of the
Framingham Study,
studied untreated blood pressure and cognitive functioning [18].
Though the study was
longitudinal, the researchers did not measure dementia, but only
cognitive functioning.
Glynn et al and Kilander et al also measured blood
pressure and
cognitive
functioning longitudinally and were excluded from the present study because
dementia was not
diagnosed [2,19]. Skoog et al conducted a -year longitudinal study
of blood pressure and dementia [20].
Their research was excluded because the data
collected began at age 70 and was
grouped in a manner that did not lend itself to metaanalysis.
Data was then extracted and placed in
2x2 tables representing dementia and
hypertension, dementia and no
hypertension, no dementia and hypertension and no
dementia and no hypertension. The odds
ratios and the 95% confidence interval were
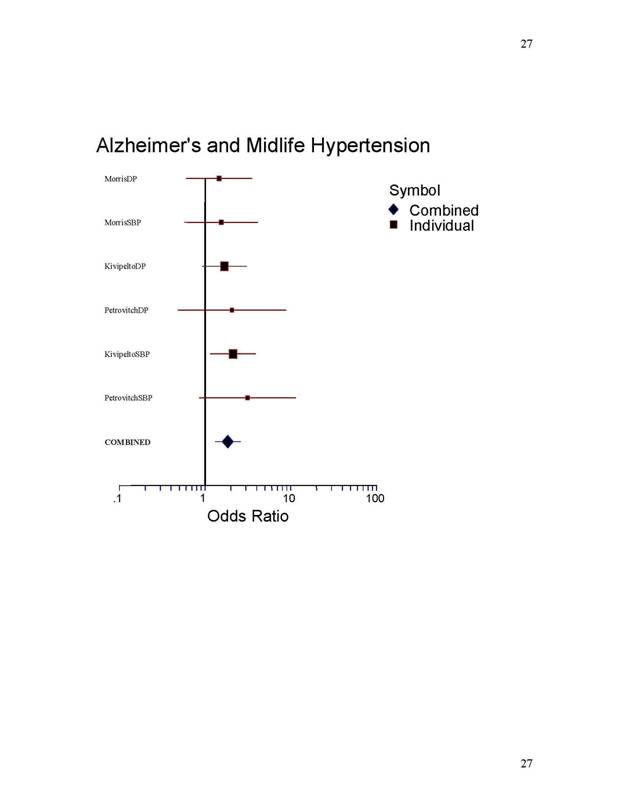
calculated using the Peto method and a forest plot was drawn (Figure 1,2) [3].
NCSS
Number Croncher
Statistical System, Kagsville Utah was utilized for
the analysis and plot.
RESULTS
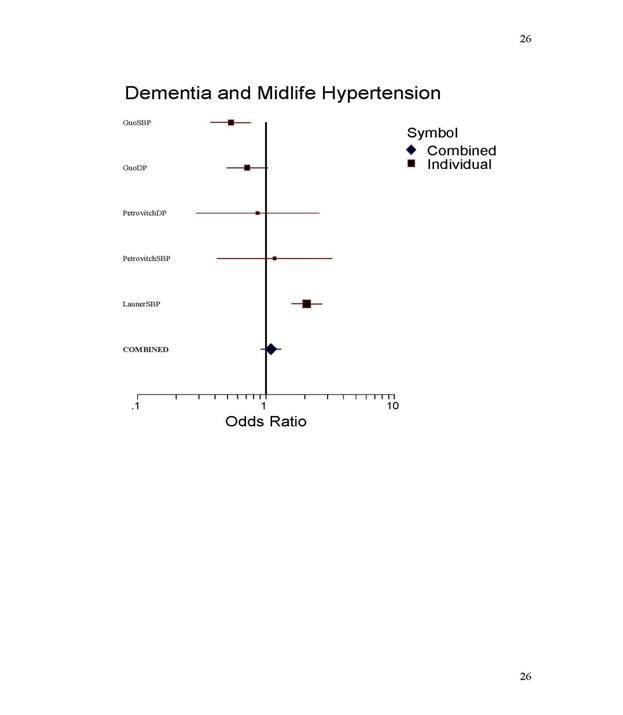
The forest plots (Figure 1,2) and
Table 1 and 2 show the statistical results of the
meta-analysis. The grouping of
dementia includes AD, vascular dementia and mixed
dementia. An odds ratio of 1.0
suggests that hypertension and dementia have no
relationship. An odds ratio of less
than one indicates that midlife hypertension and
dementia occur together less than what
one would expect by chance alone. An odds ratio
of more than one suggests that midlife
hypertension and dementia occur together more
often than would be expected by chance
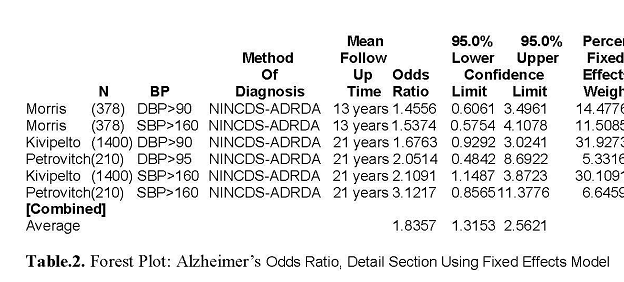
alone. The combined odds ratio of 5 dementia
studies
was 1.09. All AD studies of both elevated midlife diastolic (>90torr) and
elevated systolic (>160 torr) are
greater than one with a combined odds ratio of 1.84.
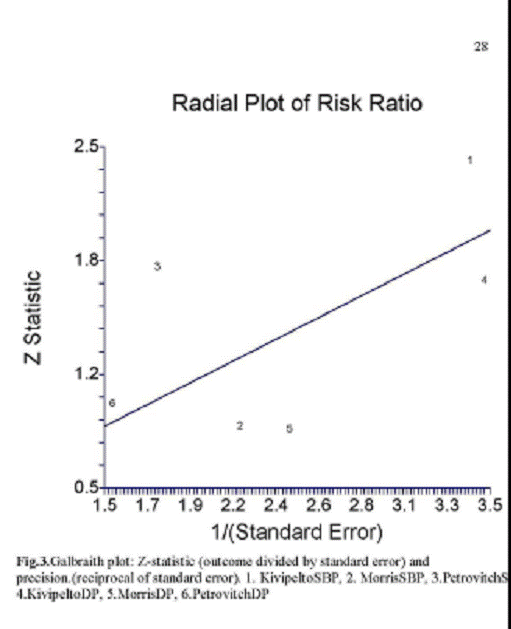
The Galbraith plot (Fig.3.) as a
estimate of publication bias does not show asymmetry
supporting the position that
unreported studies would not alter the overall finding [21].
DISCUSSION
Drawing from the
information in the forest plot and the odds ratios, it appears that
AD, but not dementia
in general, is related to midlife hypertension. This is interesting
considering the fact
that vascular dementias, of specifically a vascular origin are included
in the general
dementia category. As other causes of dementia are ruled out in the
diagnosis of AD,
midlife hypertension seems to be more and more a factor. These results
are highly
suggestive of a vascular mechanism in AD, of which hypertension may be a
precursor.
The forest plot may be somewhat
incomplete, as any unpublished data was not
included in this
analysis. Also, it is quite possible that negative results have not been
submitted for
publication. This is true with the study by Launer et
al. [4]. The raw data
was available for
their positive correlation with SBP, but not for DBP where no
correlation was
found [4]. Mean follow-up times differ between each study making it
difficult to
generalize the outcomes to a certain age or time period considered
"midlife."
With the study by
Guo et ai, follow up time was only 40.5 months [17]. This is a very
short time compared
with 15-20 years in the other studies. Guo's study demonstrates the
finding that blood
pressure tends to drop in the few years before dementia onset [17]. The
inclusion criteria
may have been too lax or too strict. The need for data to be presented in
a certain fashion in
the studies used may also limit this analysis, as one relevant paper
was excluded because
the raw data could not be extracted [20]. While the Skoog et al
data (n~94)
does not lend itself to this META
analysis, those who developed dementia
with the DSM-III-R
criteria at age 79-85 had significantly higher SBP at age 70 and
higher DBP at age 70-85 than those who
did not develop dementia [20]. These authors
also found elevated DBP associated
with AD at 79-85 and this elevated BP correlated
with white matter lesions in a subset
of 15 patients who underwent CT scans.
While the included five studies do not
allow us to pin down the precise age in
which hypertension becomes a factor,
Posner et al does suggest that after age 65
hypertension by itself is no longer a
factor [22].
The
suggestion that mean pressure is a key factor cannot be supported nor can it
be ruled out by these odds ratios. The
Diastolic odds ratios are no greater than the
systolic values.
AD as a slow continuously progressive
disease would suggest failure in fine tuning
of control systems not complete chaos
in that system. Certainly renewal and
repair must be under consideration.
Cerebral hypo perfusion and cognitive decline has
been reported in a number of papers
and has been reviewed [23,24]. Vessel
characteristics may take up to 6 years
to return to normal after hypertension even though
B.P. has normalized so reduced local
flow and abnormalities of vasomotion within
endothelial cell shape change may
contribute to the hypo perfusion. The three major
cerebral arteries ofthe
brain are end arteries without significant collateral circulation
[25]. After an early period of midlife
hypertension, failure to maintain proper perfusion
pressure would lead to cerebral hypo
perfusion, cortical infarcts, and rapid decline in the
AD patient if increased vascular
resistance and disrupted flow control continued. Any
clear explanation for the specific
anatomical regions and Braak progression, however is
not understood [26]. The critically
attained threshold of cerebral hypoperfusion (CATCH
hypothesis) has been suggested for
neurodegeneration [24,27,28] However beta-amyloid
(A beta) peptide increase in the
hippocampus and the enterorhinal cortex would
increase
local vascular resistance in cerebral
micro-vessels and maintain or create a profound
local hypoperfusion possibly
explaining while these regions that develop high A beta
peptide, would incur greater local
ischemia and continued endothelial remodeling
failures. Endothelial cells (in tissue
culture) live about as long as a RBC 120-130 days, so
a progressive inept replacement by
circulating fibroblasts from bone marrow may create a
continuous deterioration in metabolic
function in these memory active regions.
Instrument describing Dementia
It should be noted that
the instruments used to define dementia are not without
controversy [29]. Some challenge the
tools used to evaluate dementia presented in the
studies analyzed arguing that
"executive function" among others are primary and
diminish the value of "memory
mechanisms" as key [30]. It is
interesting to speculate a
reduction in the variance by using a more
tightly defined progression of regional PHF-tau
pathology in sync with serial blood
pressure changes in a model linking the clinical
presentation of dementia with anatomy
[30]. Function and anatomy almost never allow
perfect causal links but strong arguments
can be made for resolutions between Braak and
Braak
and Royall and others in pathological classification. Why can percent of
arterial
stenosis
explain 25% of the variance in Braak Stage, 36% of
the variance in CERAD
Neural Plaque Score, and 22% of the
variance in white matter score in a Sporadic AD
population [14]7
The evidence for low glucose
availability in the Entorhinal cortex and
hippocampus is supported by studies
that raise plasma insulin through intravenous
infusion while keeping plasma glucose
at fasting levels. This gave striking memory
enhancement which suggests that neuroendocrine factors are quite
important here [31].
How insulin is kept
from these regions is a puzzle but normal endothelial function is a
major factor in
insulin resistance, and diabetes mellitus was associated with lower
cognitive function
where diabetes gave a 65% increase in risk (hazard ratio 1.65) of AD
compared with those
without diabetes [32,33]. It is clear however that diabetes mellitus
clusters with other
vascular risk factors in AD [11]. While some claim a direct correlation
between
plasma insulin levels and resting blood pressure [34], more recently Jan 2020,
patterns of reduced glucose metabolism are often seen in brain scans of patients with
Alzheimer disease and other dementias. Now, a growing body of evidence suggests
that glucose hypometabolism may be more than just a biomarker on brain scans: it
may be a key player in dementia pathology.[35]
Others have found diastolic blood pressure
during exercise was higher in hypertension-
prone
and insulin-resistant patients which suggests a better measure to demonstrate
this
relationship
[36]. Possible Information present in the midlife Hypertension finding.
A
vascular elevated pressure sign that appears early then essentially goes away
in
many patients would appear to be an enigma. However this
strange hypertension may be
an explanation that supports the hypothesis that
progenitor endothelial cell involvement is
seminal in this dementia. The endothelial progenitor
cell dysfunction in hypertension has
been reviewed [37]. The unique two layer embryology of
the brain and the kidney, in
which vascular supply must come from outside these
structures, together with the
description of endothelial progenitor cells (EPC) in the
circulating blood points to
neovascularization occurring in early AD [38-39]. This
points to a possible link between
renal hypertension mechanisms and hypertensive brain
tissue changes. Endothelial cells
must be repaired and renewed, with endothelial protein
turnovers in the brain (perhaps as
rapid as every 2 weeks) imply extremely dynamic renewal
mechanisms. EPC
proliferation, migration, and adhesion as well as in
vitro vasculogenesis has been found
impaired due to elevated homocystine,
total cholesterol, LDL cholesterol, and C-reactive
Protein [40,41]. In addition the high angiotensin II
levels present during hypertension
might indicate a control system overdriven to potentate
VEGF-induced EPC proliferation
back into homeostatic conditions. It is not unreasonable
to picture elevated local vascular
resistance, metabolic abnormalities, and proinflammatory
cytokines in patients that
progress from Mild Cognitive Impairment to AD [42]. A
reasonable supposition that
would explain abnormal skin fibroblast metabolism and
perfusion in AD, would be
abnormal fibroblast progenitor endothelial cell function
[43,44]. The case can be made
that the midlife hypertension may drive or represent
this abnormality early in the
developing AD. Also the CV-19 attack virus would so
alter the endothelium as to
accelerate the whole process because of the spike
attachment to the ACE-2 receptors
in the vascular endothelium when the blood-brain barrier
was compromised.
Loss of
Hypertension symptom with severity
Studies of older patients and autopsy studies are likely
to miss signs/symptoms of
hypertension. It is accepted that in severe Braak
States IV and V the elevated pressures
have disappeared or have fallen low at late onset but the
tortuous small vessels and
heterogeneity of brain blood flow is still present [22].
Consequences of hypo perfusion: A-beta peptides
Studies in gerbil forebrain ischemia and rat chronic
(reduced 25%) hypo perfusion
Of the hippocampus has been shown sufficient to trigger
amyloid precursor protein (APP)
cleavage into A-beta peptides [45]. Supporting the idea
that atherosclerotic occlusion is
an important factor in pathogenesis of some sporadic AD
[46]. Ischemia temporarily
induced amyloid peptide over-expression in reactive
astrocytes and this over-expression
peaked at day 7 and 6 months [47]. These studies support
the cerebrovasculature as a
clinically relevant site of AD which contributes to neuro-degeneration [48].
A-beta is known to induce transendothelial
migration of monocytes/microglia in
culture that could be inhibited by the putative A-beta
receptor for advanced glycation end
products (RAGE) suggesting peripheral blood
monocytes/microglia would accumulate in
the brain of AD patients. And is a logical explanation
for the low grade inflammation and
periventricular white matter lesions usually present.
This migration is shown likely
reduced in normal brain vascular endothelium. By
injecting antibodies to A-beta directly
in the brain of mice genetically engineered to make
excess amyloid and tau found the
time constant of amyloid plaques and tangles is
remarkably short [49]. Three
days post
injection, amyloid plaques were gone. Two more days and
tau tangles were gone showing
that plaques promote tangles. If tau is allowed to add
phosphate groups it could not be
removed [49]. These
short time constants force one to search for a prolonged
environmental challenge to the genetic environment. A
stretch of midlife hypertension or
perhaps a very low B.P. might possibly be responsible [11].
Evidence of Hippocampal Atrophy with Hypertension
Further support of these findings can be found in a
recent midlife blood pressure
study of Japanese-American men in the same population
presented in the Launer et al
study relating the risk of hippocampal atrophy [50].
Untreated hypertensives had a
significantly increased risk for hippocampal atrophy
(lowest quartile of hippocampal
volume). The volume of the CAl
field of the hippocampus in hypertensive rats was
reduced but could be reversed with a calcium channel
blocker [51]. Midlife changes in
estrogen and progesterone can demonstrate real vascular
effects and these sex hormones
may directly effect calcium
channels [52]. It
is possible that hypertension that
responds to
calcium channel blockers at midlife identifies a special
subset of treatable early
Alzheimer patients.
Possibility
that a population subset is present
There may be different initiating factors in these AD
patients. An initiating
factor
without the genetic environment would not result in a
sustained disease. While fifty or
more genes may be implicated in dementia, hypertension
coupled with sex hormone
changes of midlife create powerful forcing functions for
altering the genetic
environment. There are many known changes in the
hypertensive brain. However the
demand that the initiating factor(s) for AD are the same
as the sustaining factors is not
necessary.
CONCLUSION
It seams
there is an association between AD and hypertension but why this
association exists
is not clear. Some may choose to continue to reject the idea that AD
may have a vascular
origin. While there are a number of AD patients that do not have
classical
hypertension, there seems to be a real interaction between AD and hypertension.
It is now understood
that hypo-glucose metabolism is occurring as is insulin dysfunction
meaning reduced metabolic capacity mixed with
reduced blood flow is underlying the
dementia. A clinical trial where hypertension
is identified and treated would clarify with greater
certainty what
removal of the hypertensive environment would mean for the development
of AD while AD is in
the earliest stages would be helpful. Is it possible that mis-regulation
of APP proteolytic
processing or RAGE removal of A-BETA is established by
a bout of midlife
hypertension. The possibility exists that AD has vascular, metabolic, and
neurological
precursors and the element that ties them together is endothelial
damage/repair rates and their interaction
with folding missense for A beta misfo1ding in
an ischemic or hypo-perfusion
environment of mid1ife hypertension.
REFERENCES
1 Crentsil,
V: The Pharmacogenomics of Alzheimer's disease.
Ageing Research
Reviews 2004;3: 153-69.
2 Glynn RJ, Beckett LA, Hebert LE,
Morris MC, Scherr PA, Evans DA: Current
and remote blood pressure and
cognitive decline. JAMA 1999;281:438-45.
3 Sutton AJ, Abrams KR, Jones DR,
Sheldon TA Song F: Methods for MetaAnalysis
in Medical Research. West Sussex: John
Wiley & Sons, Ltd, 2000.
4 Launer LJ,
Masaki K, Petrovich H, Foley D, Havlik
RJ: The association between
midlife blood pressure levels and
late-life cognitive function. JAMA
1995;274: 1846-51.
5 Petrovich
H, White LR, Izmirilian G, Ross GW,Bennett DA, Evans DA: Midlife
blood pressure and neuritic
plaques, neurofibrillary tangles, and brain weight at
death: the HAAS. Neurobiology of
Aging. 2000;21:57-62.
6 Morris MC, Scherr PA, Hebert LE,
Glynn RJ, Bennett DA, Evans DA:
Association of incident Alzheimer
disease and blood pressure measured from 13
years before to 2 years after
diagnosis in a large community study. Arch Neurol.
2001;58:1640-6.
16
7 Kivipelto
M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K,
Soininen
H, Tuomilehto J, Nissien A:
Midlife vascular risk factors and
Alzheimer's disease in later life. longitudinal, population based study: British
Medical Journal. 2001;322: 1447-51
8 de la Torre JC: Alzheimer disease as
a vascular disorder. Stroke 2002;33:1152-75.
9 Wu C, Zhou D, Wen C, Zhang L, Como
P, Qiao Y: Relationship between blood
pressure and Alzheimer'd
disease in Linxian county China. Life Sci
2003;72: 1125-33.
10 Hebert LE, Scherr PA, Bennett DA, Bienias JL, Wilson RS, Morris MC, Evans
DA: Blood Pressure and late-life cognitive
function change. Neurology 2004;
62:2021-14.
11 Skoog I, Gustafson D: Hypertension,
hypertension-clustering factors and
Alzheimer's disease. Neurol Res 2003;25:675-80.
12 Skoog I, Kalaria RN, Breteler MB: Vascular
factors and Alzheimer disease.
Alzheimer Disease and Associated
Disorders 1999;13:S106-S114.
17
13 Stweart
R: Cardiovascular factors in Alzheimer's disease. J Neurol
Neurosurg
Psychiatry; 1998;65:143-47.
14 Roher AE,
Esh C, Kekjohn TA, Kalbach W, Luehrs DC, Seward JD,
Sue LI,
Beach TG: Circle of Willis
atherosclerosis is risk factor for sporadic Alzheimer's
disease. Arterioscler
Thromb Vas Bioi 2003;23:2055-62.
15 Meyer JS, Rauch GM, Rauch RA, Haque
A, Crawford K: Cardiovascular and
other risk factors for Alzheimer's
disease and vascular dementia. Ann NY Acad
Sci 2000;903:411-23.
16 McKhann
G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM:
Clinical
diagnosis of Alzheimer's disease,
Report ofthe NINCDS-ADRDA Work Group
under the auspices ofthe
Department of Health and Human Services Task Force
on
Alzheimer's Disease: Neurology 1984;34:940-4.
17 Guo Z, Frtiglioni L, Winblad B, Viitanen M: Blood pressure and performance on
the Mini-Mental State Examination in
the very old. American Journal of
Epidemiology. 1997;145:1106-13.
18 Elias MF, WolfPA,
D'Agostino RB, Cobb J, White LR: Untreated blood
pressure
level is inversely related to
cognitive functioning: the Framingham study.
American Journal of Epidemiology 1993;138:353-64.
19 Kilander
L, Numan H, Boberg M,
Hansson L, Lithell H: Hypertension is related
to cognitive impairment: a 20-year
follow-up of999 men. Hypertension
1998;31 :780-786.
20 Skoog I, Lemfelt B, Landahl S, Palmertz B, Andreasson L, Nilsson
L, Persson G,
Oden
A, Svanborg
A: IS-year longitudinal study of blood pressure
and dementia.
Lancet 1996;347:1141-45.
21 Matthias E, Smith GD, Schneider M,
Minder C: Bias in meta-analysis detected by
a simple, graphical test. BMJ 1997;315:629-634.
22 Posner HB, Tang MX, Lauchsinger J, Lantigua, Stern Y,
Mayeux R. The
relationship of hypertension to
Alzheimer's Disease, vascular dementia, and
cognitive function. Neurology 2002;58:
1175-81.
23 Miklossy
J: Cerebral hypoperfusion induces cortical watershed
microinfarcts which
may further aggravate cognitive
decline in Alzheimer disease. Neurol Res
2003;25:605-10.
24 De Jong GI, Farkas E, Stienstra CM.
Plass JR, Keijser IN, de la
Torre JC, Luiten
PG: Cerebral hypoperfusion yields
capillary damage in the hippocampal CAl area
that correlated with spatial memory
impairment. Neuroscience 1999;91 :203-10.
25 Baldin
CF, Chambers BR: Clinical features, pathogenesis, and complete
tomographic characteristics of
internal watershed infarction. Stroke
1993;24: 1925-32.
26 Braak, H,
Braak E: Neuropathological stageing of Alzheimer-related changes.
Acta Neuropathol1991; 82: 239-259.
27 de la Torre JC: Critically attained
threshold of cerebral hypoperfusion; can it
cause Alzheimer's disease. Ann NY Acad Sci 2000;903:424-36.
28 de la Torre JC: Impaired cerebromicrovascular perfusion: summary of evidence in
support of its causality in
Alzheimer's disease. Ann NY Acad Sci 2000;21:321-30.
29 Royall DR: Back to the Future of
mental capacity Assessment. JAGS
2002;50: 1884-85.
30 Royall DR, Palmer R, Mulroy AR, Polk MJ, Roman GC, David JP, Delacourte
A:
Pathological determinants ofthe transition to clinical dementia in Alzheimer's
disease. Exp Aging Res 2002;28:
143-62.
31 Craft S, Walson
GS: Insulin and neurodegenerative disease: shared and specific
mechanisms. Lancet Neuro12004;3:l69-78.
32 Ling X, Martins RN, Racchi M, Craft S, Hamerhorst E.
Amyloid beta antagonizes
insulin promoted secretion of amyloid
beta protein precursor. J Alzheimer's
Disease 2002;4:369-74.
33 Arvanitakis
Z, Wilson RS, Bienias JL, Evans DA, Bennett DA: Diaabetes Mellitus
and risk of Alzheimer's disease and
decline in cognitive function. Arch Neurol
2004;61:661-6.
34 Sowers JR: Insulin resistance and
hypertension. Am J Physiol Heart Circ Physiol
2004;286:H1597-H1602.
35.Kuehn, Bridget M.,MSJ. In
Alzheimer Research, Glucose Metabolism
Moves to Center Stage. JAMA. Published online January 8, 2020.
doi:10.1001/jama.2019.20939
36 Andersen DB, Olsen MH, Dige-Petersen H, Ibsen H: Exercise blood pressure is
related to insulin resistance in
subjects with two hypertensive parents. Blood
Pressure 2003;12:314-18.
37 Loomans
CJ, Dao HH, van Zonneveld AJ, Rabelink
TJ: Is endothelial progenitor
cell dysfunction involved in altered
angiogenic processes in patients with
hypertension. CUff
Hypertens Rep 2004;6:51-54.
38 Murayama T, Asahara
T: Bon marrow-derived endothelial progenitor cells
for
vascular regeneration. CUff Opin Mol Ther
2002;4:395-402.
39 Burdinger
TF: Progenitor endothelial cell involvement in Alzheimer's disease.
Neurol Res 2003;25:617-24.
40 Chen JZ, Zhu JH, Wang XX, Zhu JH, Xie XD, Sun J, Shang YP, Guo XG, Dai
HM, Hu S1:
Effects ofhomocystine on number and activity of
endothelial
progenitor cells from peripheral
blood. J Mol Cell CardioI2004;36:233-9.
41 Verma L, Kuliszewski MA, Li SA, Szmitto
PE, Zucco L, Wang C, Badiwala
MV,
Minkle
DA, Weisel RD, Fedak PW,
Stewart DJ, Kutryk MJ: C-reactive protein
attenuates endothelial progenitor cell
survival, differentiation, and function.
Circulation 2004;109:2058-67.
42 Tarkowski
E, Andreasen N, Tarkowski
A, Blennow K: Intrathecal
inflammation
precedes development of Alzheimer's
disease. J Neurol Neurosurg
Psychiatry
2003;74: 1200-1203.
43 Peterson C, Goldman JE: Alterations
in calcium content and biochemical
processes in cultured skin fibroblasts
from aged and Alzheimer donors. Proc Natl
Acad
Sci 1986;83:2758-62.
44 Sims NR, Finegan
JM, Blass JP: Altered metabolic properties of cultured skin
fibroblasts in Alzheimer's disease. Ann
NeuroI1987;21:451-57.
45 Bennett SA, Pappas BA, Stevens WD,
Davidson CM, Fortin T, Chen 1: Cleavage
of amyloid precursor protein elicited
by chronic cerebral hypoperfusion.
Neurolbiol
Aging 2000;21:207-14.
46 Kalbach
W, Esh C, Castano EM, Rahman A, Kokjohn T, Luchus DC, Sue L,
Cisneros R, Geuber
F, Richardson C, Bohrmann B, Walker DG, Beach TG, Roher
AE: Atherosclerosis, vascular
amyloidosis and brain hypoperfusion in
pathogenesis ofsporadic
Alzheimer's disease. Neurol Res 2004;26:525-39.
47 Plura R:
Blood-brain barrier dysfunction and amyloid precursor protein
accumulation in microvascular
compartments following ischemia-reperfusion
brain injury with I-year survival. Acta Neurochir SuppI2003;86:
117-22.
48 Kalaria
RN, Bhatti SV,Lust
WD, Perry G: The Amyloid precursor protein in
ischemic brain injury and chronic
hypo-perfusion. Ann N. Y. Acad Sci
1993;695: 190-193.
49 Oddo S,
Billings L, Kesslak JP, Cribbson
DR, Laferla FM: Abeta
immunotherapy
leads to clearance of early, but not
late, hypophosphorylated tau aggregates via
the proteasome. Neuron 2004;43:321-32.
50 KorfES,
White LR, Scheltens P, Launer
LJ: Midlife blood pressure and the risk
of hippocampal atrophy: The Honolulu
Asia Aging Study. Hypertension
2004;44:29-34.
51 Sabbatini
M, Tomassoni D, Amenta F:
Hypertensive brain damage: comparative
evaluation of protective effect oftreatment with dihydropyridine
derivatives in
spontaneously hypertensive rats. Mech
Aging Dev. 2001;122:2085-2105.
52 Khalil RA: Sex hormones as
potential modulators ofvascular function in
hypertension. Hypertension 2005;46:249-254.
24
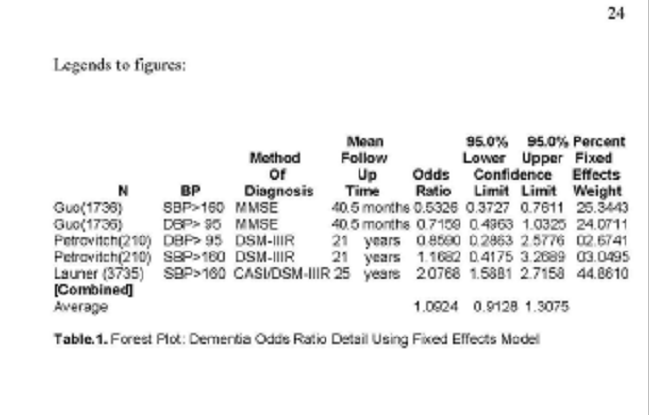
Legends to figures:
N
Guo(1736)
Guo(1736)
Petrovitch(210)
Petrovitch(210)
Launer
(3735)
[Combined]
Average
Mean 95.0% 95.0% Percent
Method Follow Lower Upper Fixed
Of Up Odds Confidence Effects
BP Diagnosis Time Ratio Limit Limit
Weight
SBP>160 MMSE 40.5 months 0.5326 0.3727 0.7611 25.3443
DBP> 95 MMSE 40.5 months 0.7159 0.4963 1.0325 24.0711
DBP> 95 DSM-IIIR 21 years 0.8590 0.2863 2.5776 02.6741
SBP>160 DSM-IIIR 21 years 1.1682 0.4175 3.2689 03.0495
SBP>160 CASI/DSM-IIIR 25 years 2.0768 1.5881 2.7158
44.8610
1.0924 0.91281.3075
Table.1. Forest Plot: Dementia Odds Ratio Detail Using
Fixed Effects Model
1.8357 1.3153 2.5621
N BP
Morris (378) DBP>90
Morris (378) SBP>160
Kivipelto
(1400) DBP>90
Petrovitch(210)
DBP>95
Kivipelto
(1400) SBP>160
Petrovitch(210)
SBP>160
[Combined]
Average
Method
Of
Diagnosis
NINCDS-ADRDA
NINCDS-ADRDA
NINCDS-ADRDA
NINCDS-ADRDA
NINCDS-ADRDA
NINCDS-ADRDA
Mean
Follow
Up Odds
Time Ratio
13 years 1.4556
13 years 1.5374
21 years 1.6763
21 years 2.0514
21 years 2.1091
21 years 3.1217
95.0% 95.0%
Lower Upper
Confidence
Limit Limit
0.6061 3.4961
0.5754 4.1078
0.9292 3.0241
0.4842 8.6922
1.1487 3.8723
0.856511.3776
25
Percer
Fixed
Effect!
Weigh
Forest Plot (2021) of drugs to reduce Amyloid. Aduhelm (Aducarimab) FDA approved 6/11/2021
See: https://www.bmj.com/content/bmj/372/bmj.n156/F2.large.jpg?width=800&height=600
{kind=link}